
Peer Reviewed
Hemophagocytic Lymphohistiocytosis
Authors:
Ammar ELJack, MD
Beaumont Hospital, Dearborn, Michigan
Isra Ibrahim, MD
Beaumont Hospital, Dearborn, Michigan
Alaa ElKhider, MD
University of Kentucky College of Medicine, Lexington, Kentucky
Mohamed Ali Mohamed, MD
Henry Ford Hospital, Detroit, Michigan
Citation:
ELJack A, Ibrahim I, ElKhider A, Mohamed MA. Hemophagocytic lymphohistiocytosis. Consultant. 2019;59(5):155-157.
An 18-year-old man presented to the emergency department (ED) with altered mental status and status epilepticus. He had recent history of 2 previous hospital admissions.
On the first admission, he presented with fever, lower extremity edema, and a generalized rash sparing the palms and soles. He had pancytopenia, elevated ferritin, and elevated liver enzymes, and a computed tomography (CT) scan of the abdomen showed small ascites and hepatosplenomegaly (Figure 1). Liver biopsy results showed fatty liver.
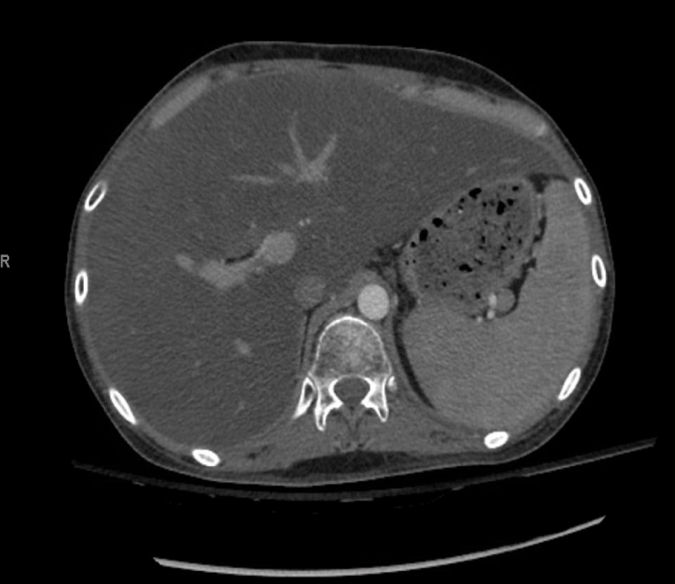
Figure 1. Abdominal CT scan showed hepatosplenomegaly and ascites.
A consulting rheumatologist diagnosed Still disease in view of the elevated ferritin level. He was started on prednisone and methotrexate and was instructed to follow up in the rheumatology clinic.
One month later, he was admitted for right eye swelling. CT of the head showed extensive cellulitis involving the scalp, face, and neck. He was started on vancomycin and azithromycin. On admission he was febrile and tachycardic, and he had circumferential erythema around his neck and mouth bleeding and an enlarged liver. Other findings of a systemic examination were unremarkable.
Laboratory tests showed pancytopenia, an extremely elevated ferritin level of 215,400 ng/mL, hypertriglyceridemia, hypofibrinogenemia, and an elevated interleukin-2 soluble receptor (soluble CD25) level. Echocardiography showed a low ejection fraction of 42% and small circumferential pericardial effusion. The results of bone marrow biopsy were compatible with a diagnosis of hemophagocytic lymphohistiocytosis (HLH). The patient was started on dexamethasone and etoposide.
Results of follow-up laboratory test done 4 weeks after discharge showed improvement in his condition. Two months later, he presented again with altered mental status and status epilepticus. T2-weighted fluid-attenuated inversion recovery (FLAIR) magnetic resonance imaging (MRI) of the brain showed multiple hyperintense lesions in the parieto-occipital lobes (Figure 2).
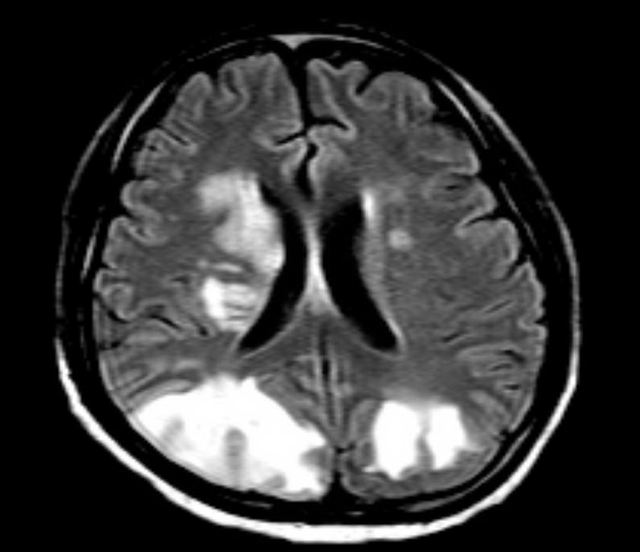
Figure 2. MRI of the brain showed multiple hyperintense lesions involving the parieto-occipital lobes.
Brian biopsy results of the left occipital lesion showed cortex and white matter involvement with few scattered macrophage infiltrates, gliosis, hemorrhage, and rare hemophagocytic cells (Figure 3).
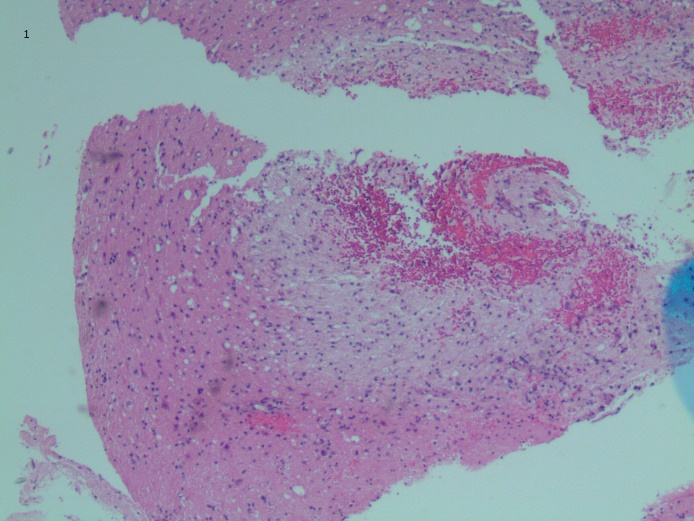
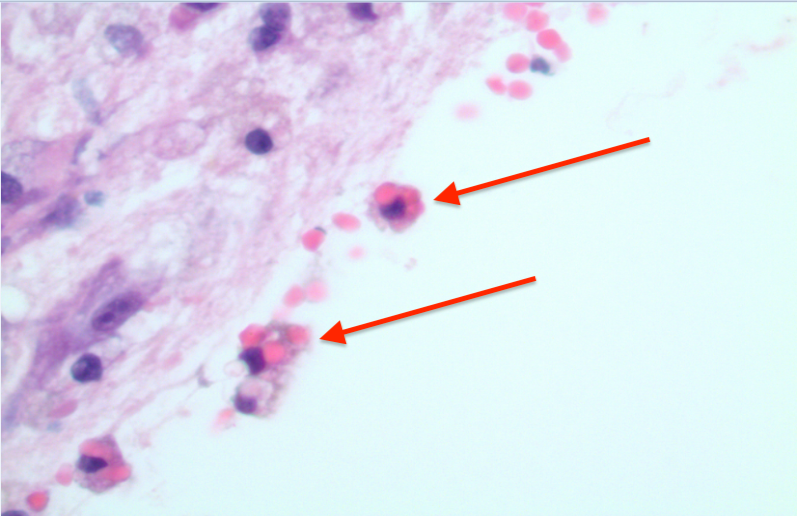
Figure 3. Brian biopsy of the left occipital lesion showed cortex and white matter involvement with few scattered macrophage infiltrates, gliosis, hemorrhage, and rare hemophagocytic cells (arrows). Hematoxylin-eosin, original magnification ×4 (top) and ×20 (bottom).
He was started on alemtuzumab and intrathecal methotrexate. He improved and was discharged to a rehabilitation center.
Three months later, he presented again to our hospital with abdominal pain, nausea, and vomiting. Amylase and lipase levels were high, in addition to very high triglycerides (8522 mg/dL). He was treated for pancreatitis secondary to hypertriglyceridemia, indicative of a relapse of HLH. His condition worsened, and he developed septic shock; despite antibiotics and vasopressors, the patient died in the hospital.
DISCUSSION
HLH is a very rare disease that is classified as either primary or secondary. The condition results from an unsuppressed immune response leading to hypercytokinemia (cytokine storm), which mimics sepsis.1 Clinically, it can present with high nonremitting fever, hepatosplenomegaly, lymphadenopathy, and central nervous system, renal, pulmonary, and cardiac involvement.2,3 Laboratory findings include pancytopenia, a high ferritin level, hypertriglyceridemia, and a high soluble CD25 level, in addition to the presence of hemophagocytic cells in bone marrow.4,5
HLH is a fatal disease, with the 17-month mortality rate in adults exceeding 50%.6 The high mortality can be tied to several factors such as a delay in diagnosis, which can be attributed to a lack of pathognomonic features and signs and symptoms that overlap with other conditions such as sepsis or bone marrow suppression. Thus, a high index of suspicion is critical for early diagnosis and survival. Urgent immunosuppressive treatment is required in these cases but can still fail to prevent complications, as in our patient’s case. Pancreatitis is not a commonly recognized complication of HLH; hypertriglyceridemia can be implicated as a reason. Patients with HLH have a high risk of relapse,7 as in our patient’s case, but no measures are available to prevent relapses.
References:
- Rosado FGN, Kim AS. Hemophagocytic lymphohistiocytosis: an update on diagnosis and pathogenesis. Am J Clin Pathol. 2013;139(6):713-727.
- Henter J-I Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131.
- Henter J-I, Göran E, Söder O, Öst Å. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paediatr Scand. 1991;80(4):428-435.
- Rivière S, Galicier L, Coppo P, et al. Reactive hemophagocytic syndrome in adults: a retrospective analysis of 162 patients. Am J Med. 2014;127(11):1118-1125.
- Arslan F, Alp S, Büyükasık Y, et al. Hemophagocytic lymphohistiocytosis in adults: low incidence of primary neoplasm as a trigger in a case series from Turkey. Mediterr J Hematol Infect Dis. 2018;10(1):e2018047.
- Brito-Zerón P, Kostov B, Moral-Moral P, et al. Prognostic factors of death in 151 adults with hemophagocytic syndrome: etiopathogenically driven analysis. Mayo Clin Proc Innov Qual Outcomes. 2018;2(3):267-27
- Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118(15):4041-4052.