
Peer Reviewed
Vogt-Koyanagi-Harada Disease
Authors:
Manasi Sejpal, MD
Extern, Peconic Bay Medical Center–Northwell Health, Riverhead, New York
Haowei Han, DO
Transitional Intern, Peconic Bay Medical Center–Northwell Health, Riverhead, New York
Lawrence M. Buono, MD
Neuro-Ophthalmology, SightMD, Hampton Bays, New York
Sandeep A. Gandhi MD
Infectious Diseases Consultant at Peconic Bay Medical Center–Northwell Health, Riverhead, New York, and Associate Professor of Clinical Medicine, New York Institute of Technology College of Osteopathic Medicine, Old Westbury, New York
Citation:
Sejpal M, Han H, Buono LM, Gandhi SA. Vogt-Koyanagi-Harada disease [published online November 25, 2019]. Consultant360.
A 26-year-old woman with no prior medical history presented to an ophthalmology clinic with retro-orbital pain, intermittent headaches, and a slowly progressive decline in vision that had begun in the right eye and had extended into the left eye over 5 days, associated with some photophobia.
The patient denied any history of sexually transmitted diseases. She had emigrated from Venezuela to the United States 4 years ago and currently worked as a gardener. There were no relieving factors, no relevant family history, and no significant zoonotic exposure. She had no history of tuberculosis or known tuberculosis exposure.
Review of systems was positive for diffuse myalgia but negative for fever, nausea, and vomiting. Her vital signs were normal. Ophthalmic examination showed a best corrected visual acuity of 20/400 in both eyes. There was bilateral diffuse hyperemia of the conjunctiva and moderate cell and flare in the anterior chamber. There were no keratic precipitates. The posterior segment examination showed mild vitreous cells with hyperemia of the optic discs and bilateral serous detachments of the macula.
Ocular coherence tomography (OCT)—a noninvasive imaging test that uses low-coherence light to produce high-resolution cross-sectional images of the retina—demonstrated significant bilateral serous fluid with neurosensory retinal detachments of the maculae (Figure 1).
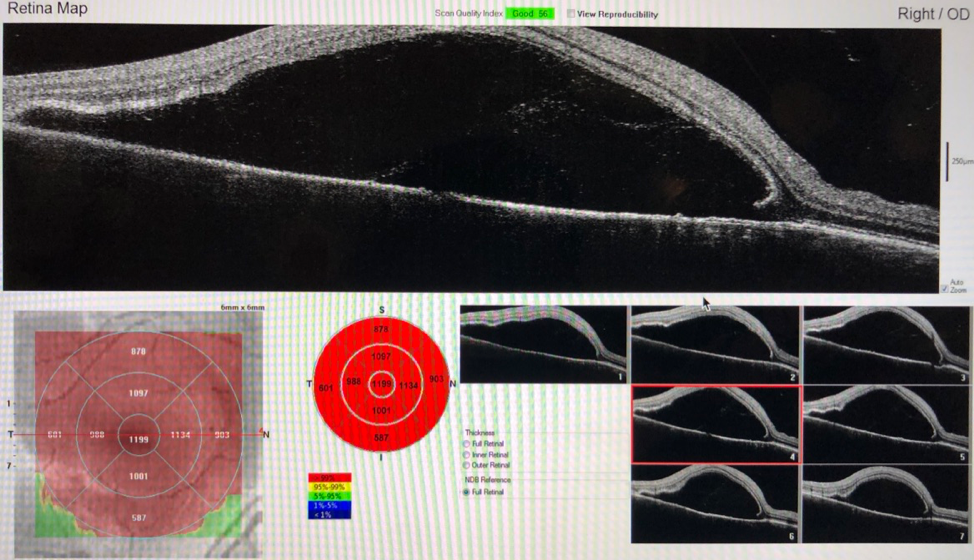
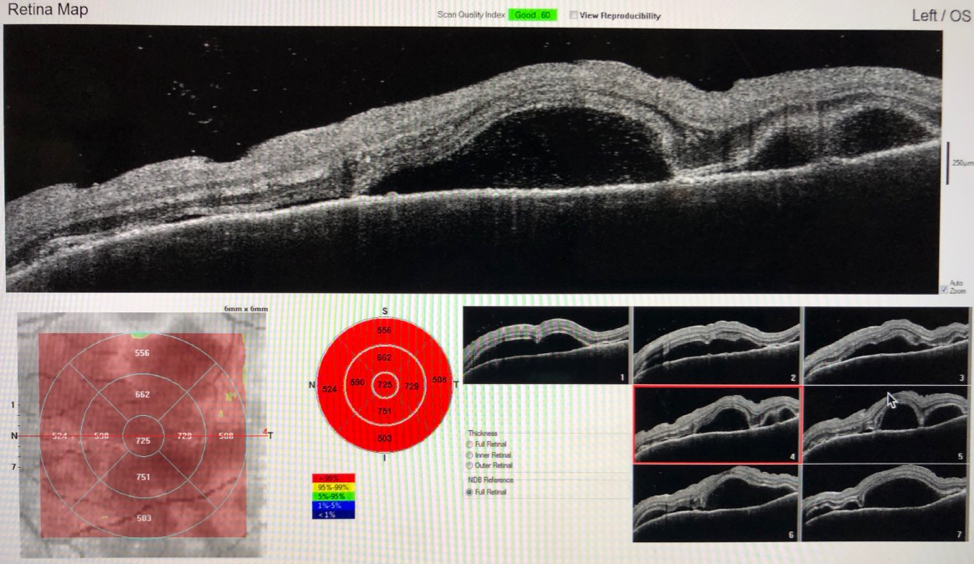
Figure 1. OCT at presentation demonstrated bilateral serous neurosensory retinal detachments of the maculae.
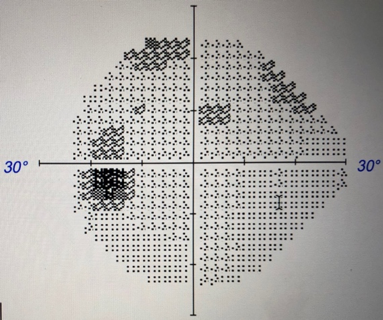
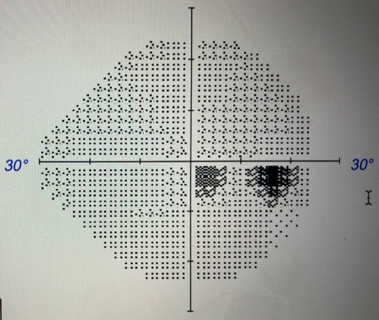
Figure 2. Humphrey automated visual field testing at presentation showed slightly enlarged anatomic blind spots, dense inferotemporal paracentral scotoma in the right eye, and scattered scotoma in the superior field in the left eye.
Neurological examination showed intact function in cranial nerves III through VII without nuchal rigidity or focal neurological deficits. Kernig and Brudzinski signs are were negative for meningitis. There were no rashes, changes in skin pigmentation, or oral or genital ulcers. The remainder of the examination findings were within normal limits.
Diagnostic tests. Results of a complete blood cell count and comprehensive metabolic panel were unremarkable. Results of a fourth-generation HIV test and serum Treponema, Toxoplasma, Bartonella, Brucella, and Lyme antibody titers were negative. The erythrocyte sedimentation rate and antinuclear antibody test results were unremarkable. Cerebrospinal fluid (CSF) analysis did not reveal leukocytes or erythrocytes. The CSF glucose level was within normal limits. The CSF albumin level was elevated. CSF and blood cultures were negative for infectious pathogens. Findings of magnetic resonance imaging (MRI) of the brain were unremarkable.
At this point, the patient was suspected to have Vogt-Koyanagi-Harada (VKH) disease. Treatment with methylprednisolone, 1 g intravenously for 3 days, was administered followed by oral prednisone, 1 mg/kg. Intravenous ceftriaxone was also given initially for empiric antimicrobial coverage prior to blood culture results. Within 3 days of intravenous corticosteroid administration, the patient’s vision and constitutional symptoms dramatically improved (Figure 3).
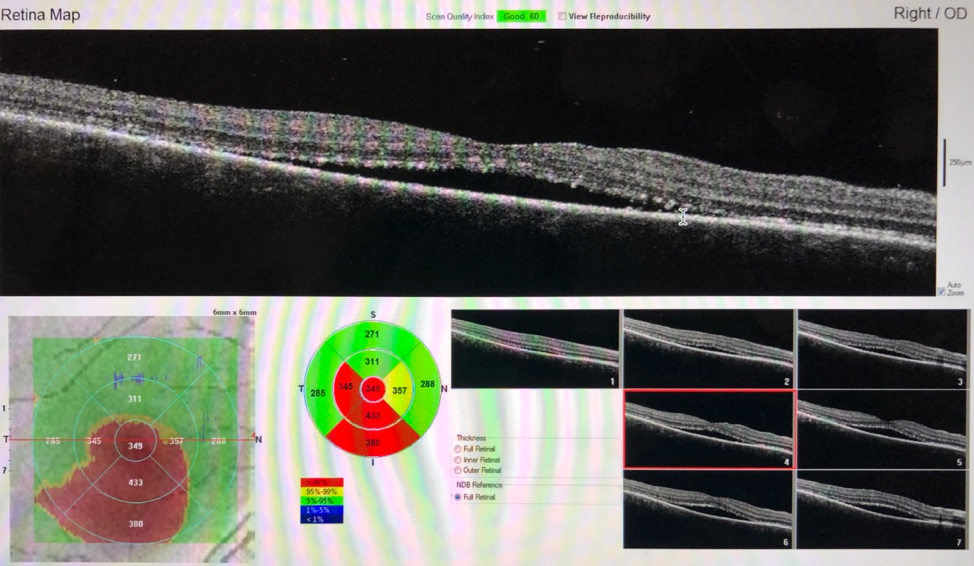
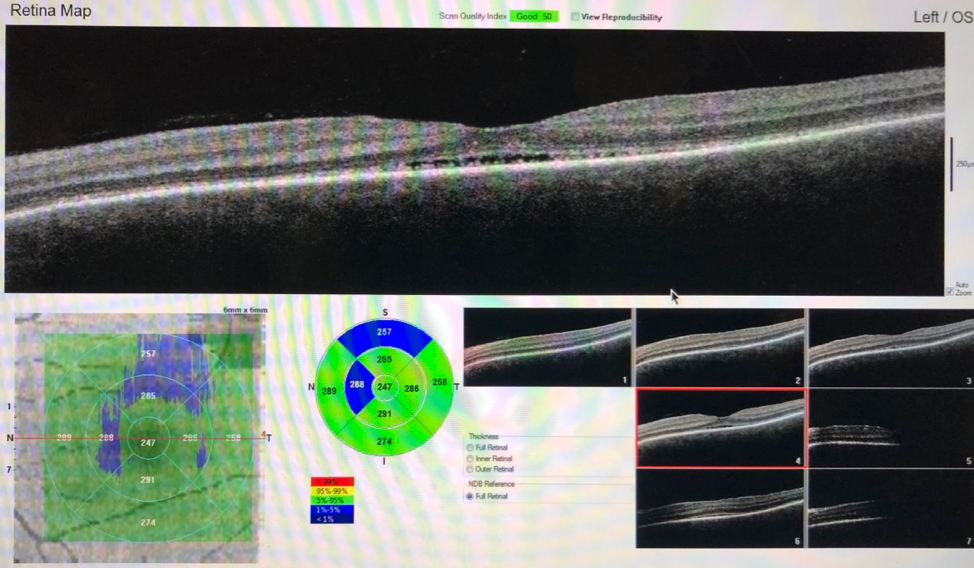
Figure 3. OCT demonstrated significant improvement of bilateral serous neurosensory retinal detachments 2 weeks after treatment with high-dose corticosteroids.
Discussion. VKH disease is a rare multisystem disease characterized by bilateral granulomatous panuveitis with unknown etiology.1 Theoretically, VKH occurs more frequently in individuals with pigmented skin, such Asians, Middle Easterners, Hispanics, and Native Americans, but less commonly in African populations.2 In the United States, VKH accounts for approximately 1% to 4% of all cases of uveitis and is seen in 1.5 to 6 per 1 million patients.2 Women are more frequently affected than men, and the most common age of onset is the second to the fifth decade; however, children and the elderly also may be affected.2
VKH disease has been described as a uveo-meningeal syndrome, since it presents with both intraocular inflammation and meningitis symptoms.3 The widely accepted theory is that it is a T-lymphocyte–mediated autoimmune reaction against melanocytes, melanin, and retinal pigmented epithelium.
Patients often present with nonspecific symptoms that resemble those of viral infections, with or without meningeal signs, for 3 to 5 days during the early course of the disease.3 Later on, during the uveitic stage, patients may begin to experience blurred visual acuity in both eyes accompanied by photophobia and ocular pain.3 Nevertheless, one important clue is that in 94% of patients, VKH disease will start with one eye and progress to bilateral involvement in 2 weeks.1
If left untreated, patients will develop serous retinal detachment and panuveitis. Neurologic and dermatologic complications will occur during the chronic stage.4 VKH also has been documented to present similarly to aseptic meningitis—notably, with headaches, focal neurologic signs, and encephalitis.5
Extraocular involvement mainly encompasses the skin, inner ears, and central nervous system (CNS). CNS manifestations can include confusion, neck stiffness, focal neurologic deficits, acute transverse myelitis, and cerebral vasculitis.2 CSF pleocytosis is seen in more than 80% of all cases.2 Inner ear involvement can include dysacusis, tinnitus, vertigo, and hearing loss, but vestibular dysfunction is uncommon.2 Integumentary involvement includes vitiligo, alopecia, and poliosis of the eyebrows, scalp, and lashes. Rarely, VKH has been reported in association with cutaneous malignant melanoma, Crohn disease, and polycystic ovary syndrome.2
VKH can be classified as probable (ocular involvement without extraocular involvement), incomplete (ocular involvement with neurologic or dermatologic involvement) or complete (ocular, neurologic, and dermatologic involvement).4 Clinical history and presentation should eliminate other causes of aseptic meningitis.5 A few additional diagnostic tests may support the diagnosis of VKH, such as lumbar puncture showing pleocytosis early in the course, fluorescein angiography, B-scan ultrasonography, and MRI, the latter of which may show diffuse choroidal thickening with scleral sparing. However, VKH remains a diagnosis of exclusion.3,6,7
The differential diagnosis of VKH includes autoimmune and malignant etiologies such as cat-scratch disease, tuberculosis, HIV infection, syphilis, sarcoidosis, lupus choroidopathy, posterior scleritis, and ocular lymphoma.5,7 Ocular complications of VKH include cataracts, retinal detachment, glaucoma, vitreous hemorrhage, and hypotony.2
The preferred treatment of VKH is high-dose intravenous corticosteroids for 3 days followed by a tapered dose of oral corticosteroids and topical corticosteroids. The duration of therapy is generally 6 months to prevent recurrence.8 Other immunosuppressive therapies include azathioprine and cyclosporine. Interestingly, tumor necrosis factor-α (TNF-α) inhibitors are also used as treatment, since TNF-α is a crucial cytokine in the formation of inflammatory granuloma.6,8 Initiating systemic corticosteroids in the first 2 weeks of disease onset leads to resolution of inflammation in 3 to 6 months, whereas a delay in treatment prolongs resolution and increases the risk of permanent vision loss.9
Conclusion. VKH should be included in the differential diagnosis in patients presenting with meningitis and progressive uveitis. Risk factors include certain ethnic groups, female gender, and changes in skin pigmentation. Patients often respond well to high-dose intravenous corticosteroids. Other immunosuppressive therapies such as azathioprine and cyclosporine may be considered in cases refractory to corticosteroid use alone.
REFERENCES:
- Salcedo HR. Vogt-Koyanagi-Harada disease. American Academy of Ophthalmology Eye Wiki. https://eyewiki.aao.org/Vogt-Koyanagi-Harada_Disease. Updated October 22, 2019. Accessed November 25, 2019.
- Lavezzo MM, Sakata VM, Morita C, et al. Vogt-Koyanagi-Harada disease: a review of a rare autoimmune disease targeting antigen of melanocytes. Orphanet J Rare Dis. 2016;11:29. doi:10.1186/s13023-016-0412-4.
- Ray R, Foroozan R. Uveo-meningeal syndromes. Int Ophthalmol Clin. 2007;47(4):131-149, x.
- Vogt-Koyanagi-Harada disease. National Organization for Rare Disorders Rare Disease Database. https://rarediseases.org/rare-diseases/vogt-koyanagi-harada-disease/. Accessed November 25, 2019.
- Sheriff F, Narayanan N, Huttner AJ, Baehring JM. Vogt-Koyanagi-Harada syndrome: a novel case and brief review of focal neurologic presentations. Neurol Neuroimmunol Neuroinflamm. 2014;1(4):e49. doi:10.1212/NXI.0000000000000049.
- Mai AP, Tran C, Wilson CW, Fox AR, Boldt HC. Vogt-Koyanagi-Harada (VKH) disease. University of Iowa Health Care, Ophthalmology and Visual Sciences. EyeRounds.org. https://webeye.ophth.uiowa.edu/eyeforum/cases/284-Vogt-Koyanagi-Harada.htm. Published April 1, 2019. Accessed November 25, 2019.
- Vogt-Koyanagi-Harada syndrome (VKH). American Journal of Neuroradiology. http://www.ajnr.org/ajnr-case-collections-diagnosis/vogt-koyanagi-harada-syndrome-vkh. December 6, 2018. Accessed November 12, 2019.
- Lodhi SA, Reddy JL, Peram V. Clinical spectrum and management options in Vogt-Koyanagi-Harada disease. Clin Ophthalmol. 2017;11:1399-1406.
- Reiff A. Clinical presentation, management and long term outcome of pars planitis (PP), panuveitis (PU) and Vogt-Koyanagi-Harada disease VKH in children and adolescents [published online October 23, 2019]. Arthritis Care Res (Hoboken). doi:10.1002/acr.24056.