
Peer Reviewed
Hereditary Hemorrhagic Telangiectasia as a Cause of Portal Hypertension
AFFILIATIONS:
1Department of Internal Medicine, University of Toledo, Toledo, OH
2Department of Gastroenterology, University of Toledo, Toledo, OH
CITATION:
Farrow D, Ramadugu A, Burlen J, Hassan M. Hereditary hemorrhagic telangiectasia as a cause of portal hypertension. Consultant. 2023;63(8):e7. doi:10.25270/con.2023.08.000006.
Received July 12, 2022. Accepted November 28, 2022. Published online August 10, 2023.
DISCLOSURES:
The authors report no relevant financial relationships.
ACKNOWLEDGEMENTS
None.
CORRESPONDENCE:
David Farrow, MD, 2100 W Central Ave, Toledo, OH 43606 (david.farrow@utoledo.edu)
A 60-year-old woman presented to an outside hospital with hematochezia, hypotension, 4 days of abdominal pain, and shortness of breath.
History. Her history was significant for type 2 diabetes, deep vein thrombosis, hypertension, and hypothyroidism. The patient had no known history of liver disease but reported non-steroidal anti-inflammatory drug use three times daily for the last 3 years for chronic back pain.
Diagnostic testing. At the outside facility, she was found to be anemic with a hemoglobin of 8.0 g/dL. An upper endoscopy demonstrated gastric varices, two columns of large esophageal varices with red wale sign, and a large amount of blood and clots in the stomach. The patient was stabilized and subsequently transferred to our facility for further management.
She was normotensive with mild tachycardia and tachypnea on arrival. Physical examination was significant for splenomegaly and mild abdominal distension. Her examination was negative for tenderness, shifting dullness, spider angiomata, palmar erythema, asterixis, or jaundice. Pertinent laboratory values are listed below (Table).
Table. Laboratory values on admission. | |
Laboratory Test | Value |
Hemoglobin | 9.2 g/dl |
Platelets | 237x 10^9/l |
Alkaline phosphatase | 286 u/l |
Albumin | 3.7 g/dl |
Total bilirubin | 0.8 mg/dl |
Aspartate aminotransferase | 24 u/l |
Prothrombin time | 12.7 seconds |
International normalized ratio | 1.1 |
An ultrasound was performed and showed coarsened hepatic echotexture with a nodular surface contour compatible with cirrhosis and no focal hepatic lesion. The main portal vein was patent without thrombus and showed a reversal of flow. The right hepatic artery, porta hepatis, and left, middle, and right portal veins were all within normal limits. Trace hepatic ascites was present. The gall bladder was surgically absent and the common bile duct measured 4 mm. A computed tomography (CT) angiography was performed at the outside facility and did not show a discrete lesion; however, the portal vein was attenuated during the arterial phase, suggesting a potential arterioportal fistula (APF).
At our facility, an esophagogastroduodenoscopy (EGD) was performed, which demonstrated esophageal and type I isolated gastric varices (Figures 1 and 2). Multiple clots and fresh blood were found in the fundus.

Figure 1. Esophageal varcies demonstrating red wale sign at our facility.

Figure 2. Esopghageal varcies demonstrated on endoscopy at our facility.
Differential diagnosis. Included on the differential diagnosis were conditions that cause telangiectasias or increased bleeding. Hereditary hemorrhagic telangiectasia (HHT), von Willebrand disease, CREST syndrome, chronic liver disease, and spontaneous telangiectasia development with age were all considered.
Multiple telangiectasia, especially in the lips and mucosal surfaces, best distinguishes HHT in these patients. In patient who are found to have multiple arteriovenous malformations in the liver, lungs, or brain, a diagnosis of HHT should be highly suspected as few other conditions cause multiple arteriovenous malformations of this nature.
Treatment and management. Interventional radiology (IR) specialists were then consulted for a transjugular intrahepatic portosystemic shunt (TIPS) procedure and embolization. The patient tolerated the procedure well and was discharged after routine post-procedure monitoring, with follow-up for further embolization procedures with IR specialists.
However, the patient presented to the outside hospital 10 days later for shortness of breath and leg swelling and was treated for acute heart failure. While at the facility, she had four large bloody bowel movements, prompting a second transfer to our facility for further management.
When the patient arrived at our facility, she had two further episodes of bright red rectal bleeding and reported mild generalized abdominal pain. The examination was negative for tenderness, distension, or jaundice. Pertinent laboratory values included hemoglobin 8.9 g/dl and a platelet count of 170 x10^9/l. All further laboratory tests were within normal limits.
Following the test results, IR specialists were consulted again to evaluate the TIPS patency and for evaluation of the source of bleeding. The TIPS patency was confirmed with abdominal ultrasound, which showed patent TIPS with end-to-end flow with elevated flow velocities in the mid-to-distal TIPS stent. We believed this was secondary to the known hepatic artery to portal venous left-to-right shunting. The abdominal ultrasound also showed a pulsatile hepatoportal vein, suggestive of right heart failure.
A hepatic arteriogram was performed (Figure 3), which demonstrated multiple peripheral hepatic artery to portal vein malformations/fistulas, the largest of which was coiled and treated using particle embolization. The treatment of remaining the arteriovenous malformations was planned in a staged fashion to avoid hepatic ischemia given the diffuse nature of the malformations.
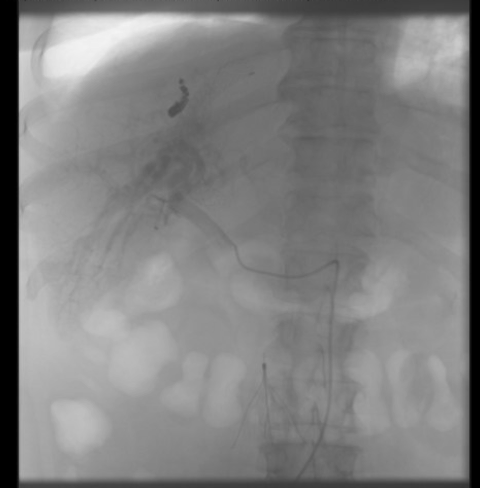
Figure 3. Hepatic arteriogram demonstrating diffuse contrast shunting throughout the liver, suggesting diffuse malformation.
Outcome and Follow-up. Upon obtaining further history from the patient, she endorsed a personal history of recurrent epistaxis, in addition to a family history of epistaxis in her mother and siblings. Based on her history and clinical picture, the patient was found to meet three of the Curacao Diagnostic Criteria for hereditary hemorrhagic telangiectasia. Therefore, she was referred for genetic counseling upon discharge. However, she did not wish to pursue further evaluation and further testing was not done.
A follow-up EGD was performed 2.5 months later due to concerns for ongoing intermittent bleeding secondary to a drop in hemoglobin noted by her primary care provider. The EGD showed large esophageal varices that were successfully treated with band ligation. No further source of bleeding was found during the procedure.
Discussion. Hereditary hemorrhagic telangiectasia (HHT), also known as Osler Webber Rendu syndrome, is a rare autosomal dominant syndrome with an estimated incidence of 1-2 per 10,000 people which causes malformation of blood vessels in the skin and organs.1 This commonly presents as excessive bleeding from mucosal surfaces, including the nose as seen in our patient. Vascular malformations are known to occur in the visceral organs in HHT.1
Our patient was found to have HHT through the finding of an APF, a common phenomenon of HHT with hepatic involvement. Upon evaluation of her APF, which can be evaluated via ultrasound or triphasic CT scan, a discrete lesion was not found. Our patient with HHT did not have a single discreet malformation but instead demonstrated a more diffuse pattern of shunting through malformed sinusoids.
Thorough assessment of personal and family medical history is essential to diagnose HHT. The Curacao Diagnostic Criteria is used to evaluate and diagnose HHT.2 The criteria includes: recurrent and spontaneous epistaxis; multiple telangiectasia of the skin, lips or oral cavity; arteriovenous (AV) malformations or telangiectasias in one or more of the internal organs; and a family history of HHT (ie, a first degree relative meets the same criteria). Meeting fewer than two of the criteria means HHT is a less likely diagnosis; meeting two makes HHT a possible diagnosis; and meeting three or more is considered a definite diagnosis of HHT. Our patient met three of the diagnostic criteria with recurrent epistaxis, AV malformations of internal organs, and a positive family history.
Patients with HHT often require coordinated multidisciplinary care from a variety of specialties because AV malformations caused by HHT can be variable not only in location but also in morbidity. Often, the care of IR specialists is pivotal in treatment. Because patient presentations can be variable, care teams should reflect individual patient manifestations. Telangiectasias in the lungs, for example, can cause direct right-to-left shunting of blood and have been associated with brain abscesses and embolic strokes.3
The standard of treatment in the past was a surgical ligation of the artery, which has lost favor due to recent advancements in IR techniques. A TIPS procedure can help alleviate hypertension in the portal system. For patients with HHT without a discrete single lesion, a more long-term approach with TIPS paired with pruning of the hepatic artery and vasculature is required. The malformed hepatic sinusoids allow diffuse shunting, making treatment during a single procedure difficult.4 Staged procedures with coiling ensure that hepatic ischemia does not occur due to a drastic reduction in blood supply to the liver.5
Before proceeding with a TIPS procedure, a baseline cardiac evaluation with echocardiogram should be performed to establish cardiac function and screen for any pre-existing conditions. TIPS placement effectively creates a left-to-right shunt and can potentially create or exacerbate any pre-existing cardiac conditions. As many as 20% of patients who undergo TIPS placement suffer from cardiac decompensation within 1 year of placement.6 A thorough discussion of risks and benefits is essential to facilitate understanding and compliance with therapy. Patients who receive TIPS should be closely monitored for signs and symptoms of heart failure in the postoperative period.
Conclusion. Patients with bleeding from a gastrointestinal source should prompt a thorough history with careful questioning not only about patient's previous history of liver disease and gastrointestinal bleeding, but also careful discussion of any history of bleeding, which may prompt consideration of hematologic disorders. Family history should also be taken to screen for information on risks for related conditions. Any family history of bleeding should prompt consideration of inherited conditions such as HHT.
- Garcia-Tsao G. Liver involvement in hereditary hemorrhagic telangiectasia (HHT). J Hepatol. 2007;46(3):499-507. doi:10.1016/j.jhep.2006.12.008.
- Faughnan ME, Mager JJ, Hetts SW, et al. Second international guidelines for the diagnosis and management of hereditary hemorrhagic telangiectasia. Ann Int Med. 2020;173(12);989-1001. doi:10.7326/M20-1443.
- Sabbà C, Pasculli G, Cirulli A, et al. Hereditary hemorrhagic teleangiectasia (Rendu-Osler-Weber disease). Minerva Cardioangiol. 2002 ;50(3):221-238.
- Chavan A, Galanski M, Wagner S, et al. Hereditary hemorrhagic telangiectasia: effective protocol for embolization of hepatic vascular malformations-experience in five patients. Radiology. 1998;209(3):735-739. doi:10.1148/radiology.209.3.984467.
- Weinstein J, Lao J, Gonsalves C, et al. Staged embolization of a complex hepatic arterioportal fistula. J Vasc Interv Radiol. 2016;3(27):S208. doi:10.1016/j.jvvir.2015.12.537
- Billey C, Billet S, Robic MA, et al. A prospective study identifying predictive factors of cardiac decompensation after transjugular intrahepatic portosystemic shunt: the toulouse algorithm. Hepatology. 2019;70(6):1928-1941. doi:10.1002/hep.30934.